- Scarpa M. et al. (2023). Acid sphingomyelinase deficiency (ASMD): addressing knowledge gaps in unmet needs and patient journey in Italy-a Delphi consensus. Intern Emerg Med. 2023 Apr;18(3):831-842. doi: 10.1007/s11739-023-03238-3. Epub 2023 Mar 7. PMID: 36882619.
- McGovern MM. et al. (2017). Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017 Feb 23;12(1):41. doi: 10.1186/s13023-017-0572-x. PMID: 28228103; PMCID: PMC5322625.
- McGovern MM. et al. (2004). Lipid abnormalities in children with types A and B Niemann Pick disease. J Pediatr. 2004;145:77–81. doi: 10.1016/j.jpeds.2004.02.048.
- McGovern MM. et al. (2017). Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017 Sep;19(9):967-974. doi: 10.1038/gim.2017.7. Epub 2017 Apr 13. PMID: 28406489; PMCID: PMC5589980.
ASMD: a disease that never ceases to surprise
The lysosomal autosomal recessive storage disease is caused by SMPD1 mutations. What are the most important differential diagnoses?
The Neurology Blog
By Annabelle Eckert
ASMD: Lack of activity of acid sphingomyelinase
There are currently various gaps in our knowledge of ASMD (acid sphingomyelinase deficiency). A distinction is made between 3 phenotypes: Niemann-Pick disease (NPD) type A (NPD A), type B (NPD B), and the intermediate type A/B. Types A and B of ASMD used to be categorised together with type C as Niemann-Pick disease. However, it is now known that type C is not caused by a lack of acid sphingomyelinase, but in 95% of cases by a mutation in the NP-C1 gene. The impaired lipid transport in type C leads to abnormal cholesterol storage. Types A and B, on the other hand, belong to the sphingolipidoses.1-3
The various types of ASMD are characterised by a deficiency in the activity of acid sphingomyelinase. This results in the accumulation of sphingomyelin in various body tissues (liver, spleen, lungs, bones and the CNS). Patients suffer from hepatosplenomegaly, interstitial lung disease, heart disease, skeletal abnormalities, growth retardation and haematological abnormalities. They have an atherogenic lipid profile.1,2
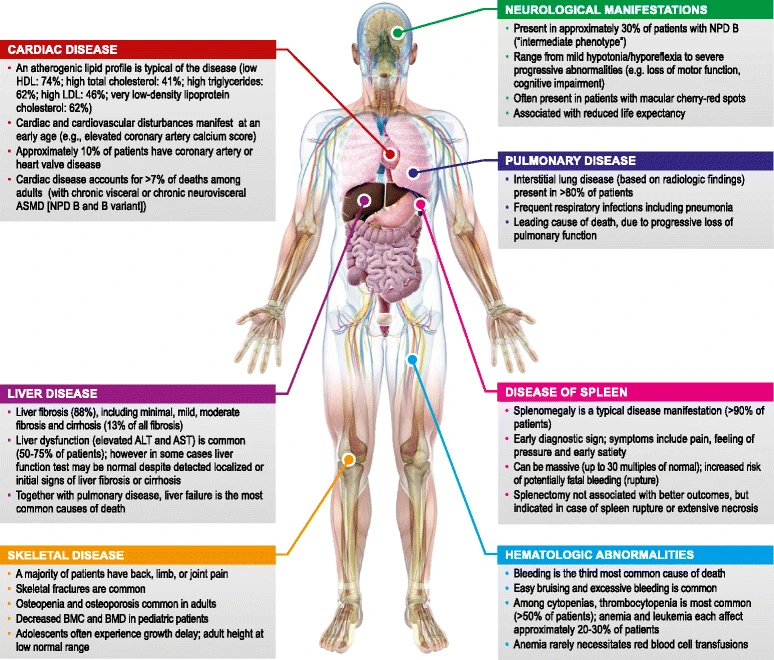
Figure 1: Compared to type A, patients with NPD B show great phenotypic heterogeneity. In this figure, McGovern MM. et al. show the different ASMD manifestations in patients with NPD B. [ALT: alanine aminotransferases; ASMD: acid sphingomyelinase deficiency; AST: aspartate aminotransferase; BMC: bone mineral content; BMD: bone mineral density; NPD: Niemann-Pick disease.]2
Half of ASMD patients suffer from haematological abnormalities
In a cross-sectional study by McGovern MM. et al. from 2004, almost half of the patients with NPD A and B had haematological abnormalities. 49% of patients suffered from bleeding episodes, with the most common bleeding event being recurrent epistaxis (29% of patients). The following significant bleeding events occurred sporadically in the aforementioned cross-sectional study:
- Excessive bleeding after tonsillectomy or adenoidectomy with blood transfusion need.
- Menorrhagia and uterine haemorrhage requiring hysterectomy.
- Subdural haematoma.
- Haematemesis.
- Haemoptysis.
- Haemothorax.
Thrombocytopenia was present in 53% of patients. 26% of patients suffered from anaemia and 21% from leucopenia.2,3
The most important differential diagnoses of ASMD
The differential diagnoses of ASMD include other storage diseases such as lysosomal acid lipase deficiency (LAL-D), Gaucher's disease, and Niemann-Pick type C. The majority of ASMD patients suffer from hepatosplenomegaly with elevated transaminases. Fibrotic remodelling of the liver and the development of liver cirrhosis can occur in the course of the disease. The most important differential diagnoses with regard to hepatosplenomegaly include primary liver diseases (e.g. cryptogenic cirrhosis, chronic hepatitis B, fatty liver and autoimmune liver changes) and malignant diseases.
Compared to other metabolic diseases such as mevalonate kinase deficiency, lysinuric protein intolerance, and transaldolase deficiency, hepatosplenomegaly is much more pronounced in ASMD. Hepatosplenomegaly is also milder in Farber disease, the mucopolysaccharidoses and the GM1 and GM2 gangliosidoses. ASMD has a typical lipid profile characterised by elevated LDL cholesterol, VLDL cholesterol and triglyceride levels. HDL cholesterol levels are significantly reduced. A similar lipid profile is observed in LALD. Patients with Gaucher disease also have low HDL cholesterol levels, but these are higher than in ASMD.1-3